Bioinformatics Cost Optimization for Computing Resources Using Nextflow (Part 1)

Many bioinformatics tools provide options to adjust the number of threads or CPU cores, which can reduce execution time with a modest increase in resource cost. But does doubling computational resources always result in processes running twice as fast? In practice, the speed-up is often less than linear, and each tool behaves differently.
In this post, I'll demonstrate how to optimize resource usage for individual tools, using a simple benchmarking workflow as a case study. The goal is to help you tune your workflow for efficient, production-ready execution.
With Nextflow—now one of the most widely used workflow managers—you gain powerful built-in features for tracing and reporting resource usage. By leveraging these capabilities, you can accurately measure CPU, memory, and run time for each step, helping you make informed optimization decisions to save both time and cost.
Getting Started with a Simple Workflow
nf-core and Nextflow have become increasingly complex. Therefore, instead of jumping directly to nf-core/rnaseq, I'll use a simple workflow to demonstrate the concepts and what we need to know first. Later, we can apply the lessons learned from this workflow to improve more complex pipelines.
I created a simple workflow using two common tools that support thread/core options.
- Tools: FastQC and fastp
- Benchmarking approach: Run tools with increasing CPU and memory allocations (2x and 3x) to observe resource consumption and execution time
- Repository: https://github.com/nttg8100/nextflow-cost-optimization
Workflow Structure
├── LICENSE
├── Makefile
├── README.md
├── benchmark.nf # Main workflow file
├── inputs
│ ├── 1_samplesheet.csv
│ └── full_samplesheet.csv
├── modules
│ ├── fastp.nf
│ └── fastqc.nf
├── nextflow.config
├── pixi.lock
├── pixi.toml
Workflow Overview
The following is a simple workflow with FastQC and fastp. It accepts a CSV file containing metadata and file paths for each sample, then performs FastQC and fastp independently.
- Aliasing strategy: Use aliases with prefix
_\<number>(e.g., FASTQC_1, FASTQC_2) to configure increasing computing resources for benchmarking
include { FASTQC as FASTQC_1 } from './modules/fastqc.nf'
include { FASTQC as FASTQC_2 } from './modules/fastqc.nf'
include { FASTQC as FASTQC_3 } from './modules/fastqc.nf'
include { FASTP as FASTP_1 } from './modules/fastp.nf'
include { FASTP as FASTP_2 } from './modules/fastp.nf'
include { FASTP as FASTP_3 } from './modules/fastp.nf'
// Parse CSV; skip header, split by comma, expand to tuple for process
Channel
.fromPath(params.input)
.splitCsv(header:true)
.map { row ->
// Compose the meta map and reads list
def meta = [ id: row.sample, strandedness: row.strandedness ]
def reads = [ row.fastq_1, row.fastq_2 ]
tuple(meta, reads)
}
.set { sample_ch }
workflow {
if (params.run_fastqc) {
// fastqc benchmarks
FASTQC_1(sample_ch)
FASTQC_2(sample_ch)
FASTQC_3(sample_ch)
}
if (params.run_fastp) {
// fastp benchmarks
FASTP_1(sample_ch,[], false, false, false)
FASTP_2(sample_ch,[], false, false, false)
FASTP_3(sample_ch,[], false, false, false)
}
}
Modules
FastQC
This module uses containers to run FastQC with configurable resources.
process FASTQC {
tag "${meta.id}"
label 'process_medium'
conda "${moduleDir}/environment.yml"
container "${ workflow.containerEngine == 'singularity' && !task.ext.singularity_pull_docker_container ?
'https://depot.galaxyproject.org/singularity/fastqc:0.12.1--hdfd78af_0' :
'biocontainers/fastqc:0.12.1--hdfd78af_0' }"
input:
tuple val(meta), path(reads)
output:
tuple val(meta), path("*.html"), emit: html
tuple val(meta), path("*.zip") , emit: zip
path "versions.yml" , emit: versions
when:
task.ext.when == null || task.ext.when
script:
def args = task.ext.args ?: ''
def prefix = task.ext.prefix ?: "${meta.id}"
// Make list of old name and new name pairs to use for renaming in the bash while loop
def old_new_pairs = reads instanceof Path || reads.size() == 1 ? [[ reads, "${prefix}.${reads.extension}" ]] : reads.withIndex().collect { entry, index -> [ entry, "${prefix}_${index + 1}.${entry.extension}" ] }
def rename_to = old_new_pairs*.join(' ').join(' ')
def renamed_files = old_new_pairs.collect{ _old_name, new_name -> new_name }.join(' ')
// The total amount of allocated RAM by FastQC equals the number of threads defined (--threads) times the amount of RAM defined (--memory)
// https://github.com/s-andrews/FastQC/blob/1faeea0412093224d7f6a07f777fad60a5650795/fastqc#L211-L222
// Dividing task.memory by task.cpus allows us to stick to the requested amount of RAM in the label
def memory_in_mb = task.memory ? task.memory.toUnit('MB') / task.cpus : null
// FastQC memory value allowed range (100 - 10000)
def fastqc_memory = memory_in_mb > 10000 ? 10000 : (memory_in_mb < 100 ? 100 : memory_in_mb)
"""
printf "%s %s\\n" ${rename_to} | while read old_name new_name; do
[ -f "\${new_name}" ] || ln -s \$old_name \$new_name
done
fastqc \\
${args} \\
--threads ${task.cpus} \\
--memory ${fastqc_memory} \\
${renamed_files}
cat <<-END_VERSIONS > versions.yml
"${task.process}":
fastqc: \$( fastqc --version | sed '/FastQC v/!d; s/.*v//' )
END_VERSIONS
"""
stub:
def prefix = task.ext.prefix ?: "${meta.id}"
"""
touch ${prefix}.html
touch ${prefix}.zip
cat <<-END_VERSIONS > versions.yml
"${task.process}":
fastqc: \$( fastqc --version | sed '/FastQC v/!d; s/.*v//' )
END_VERSIONS
"""
}
Overall, this script creates a command to run with threads and memory options. Here, memory is allocated per thread instead of total memory. If I allocate 8 threads and 16 GB RAM, it will run the command as follows:
#!/bin/bash -ue
printf "%s %s\n" SRX1603630_T1_1.fastq.gz GM12878_REP2_1.gz SRX1603630_T1_2.fastq.gz GM12878_REP2_2.gz | while read old_name new_name; do
[ -f "${new_name}" ] || ln -s $old_name $new_name
done
fastqc \
\
--threads 8 \
--memory 2048 \
GM12878_REP2_1.gz GM12878_REP2_2.gz
cat <<-END_VERSIONS > versions.yml
"FASTQC_3":
fastqc: $( fastqc --version | sed '/FastQC v/!d; s/.*v//' )
END_VERSIONS
FASTP
fastp is simpler regarding CPUs and memory. It allows direct specification without needing to recalculate memory per thread.
process FASTP {
tag "$meta.id"
label 'process_medium'
conda "${moduleDir}/environment.yml"
container "${ workflow.containerEngine == 'singularity' && !task.ext.singularity_pull_docker_container ?
'https://community-cr-prod.seqera.io/docker/registry/v2/blobs/sha256/88/889a182b8066804f4799f3808a5813ad601381a8a0e3baa4ab8d73e739b97001/data' :
'community.wave.seqera.io/library/fastp:0.24.0--62c97b06e8447690' }"
input:
tuple val(meta), path(reads)
path adapter_fasta
val discard_trimmed_pass
val save_trimmed_fail
val save_merged
output:
tuple val(meta), path('*.fastp.fastq.gz') , optional:true, emit: reads
tuple val(meta), path('*.json') , emit: json
tuple val(meta), path('*.html') , emit: html
tuple val(meta), path('*.log') , emit: log
tuple val(meta), path('*.fail.fastq.gz') , optional:true, emit: reads_fail
tuple val(meta), path('*.merged.fastq.gz'), optional:true, emit: reads_merged
path "versions.yml" , emit: versions
when:
task.ext.when == null || task.ext.when
script:
def args = task.ext.args ?: ''
def prefix = task.ext.prefix ?: "${meta.id}"
def adapter_list = adapter_fasta ? "--adapter_fasta ${adapter_fasta}" : ""
def fail_fastq = save_trimmed_fail && meta.single_end ? "--failed_out ${prefix}.fail.fastq.gz" : save_trimmed_fail && !meta.single_end ? "--failed_out ${prefix}.paired.fail.fastq.gz --unpaired1 ${prefix}_1.fail.fastq.gz --unpaired2 ${prefix}_2.fail.fastq.gz" : ''
def out_fq1 = discard_trimmed_pass ?: ( meta.single_end ? "--out1 ${prefix}.fastp.fastq.gz" : "--out1 ${prefix}_1.fastp.fastq.gz" )
def out_fq2 = discard_trimmed_pass ?: "--out2 ${prefix}_2.fastp.fastq.gz"
// Added soft-links to original fastqs for consistent naming in MultiQC
// Use single ended for interleaved. Add --interleaved_in in config.
if ( task.ext.args?.contains('--interleaved_in') ) {
"""
[ ! -f ${prefix}.fastq.gz ] && ln -sf $reads ${prefix}.fastq.gz
fastp \\
--stdout \\
--in1 ${prefix}.fastq.gz \\
--thread $task.cpus \\
--json ${prefix}.fastp.json \\
--html ${prefix}.fastp.html \\
$adapter_list \\
$fail_fastq \\
$args \\
2>| >(tee ${prefix}.fastp.log >&2) \\
| gzip -c > ${prefix}.fastp.fastq.gz
cat <<-END_VERSIONS > versions.yml
"${task.process}":
fastp: \$(fastp --version 2>&1 | sed -e "s/fastp //g")
END_VERSIONS
"""
} else if (meta.single_end) {
"""
[ ! -f ${prefix}.fastq.gz ] && ln -sf $reads ${prefix}.fastq.gz
fastp \\
--in1 ${prefix}.fastq.gz \\
$out_fq1 \\
--thread $task.cpus \\
--json ${prefix}.fastp.json \\
--html ${prefix}.fastp.html \\
$adapter_list \\
$fail_fastq \\
$args \\
2>| >(tee ${prefix}.fastp.log >&2)
cat <<-END_VERSIONS > versions.yml
"${task.process}":
fastp: \$(fastp --version 2>&1 | sed -e "s/fastp //g")
END_VERSIONS
"""
} else {
def merge_fastq = save_merged ? "-m --merged_out ${prefix}.merged.fastq.gz" : ''
"""
[ ! -f ${prefix}_1.fastq.gz ] && ln -sf ${reads[0]} ${prefix}_1.fastq.gz
[ ! -f ${prefix}_2.fastq.gz ] && ln -sf ${reads[1]} ${prefix}_2.fastq.gz
fastp \\
--in1 ${prefix}_1.fastq.gz \\
--in2 ${prefix}_2.fastq.gz \\
$out_fq1 \\
$out_fq2 \\
--json ${prefix}.fastp.json \\
--html ${prefix}.fastp.html \\
$adapter_list \\
$fail_fastq \\
$merge_fastq \\
--thread $task.cpus \\
--detect_adapter_for_pe \\
$args \\
2>| >(tee ${prefix}.fastp.log >&2)
cat <<-END_VERSIONS > versions.yml
"${task.process}":
fastp: \$(fastp --version 2>&1 | sed -e "s/fastp //g")
END_VERSIONS
"""
}
stub:
def prefix = task.ext.prefix ?: "${meta.id}"
def is_single_output = task.ext.args?.contains('--interleaved_in') || meta.single_end
def touch_reads = (discard_trimmed_pass) ? "" : (is_single_output) ? "echo '' | gzip > ${prefix}.fastp.fastq.gz" : "echo '' | gzip > ${prefix}_1.fastp.fastq.gz ; echo '' | gzip > ${prefix}_2.fastp.fastq.gz"
def touch_merged = (!is_single_output && save_merged) ? "echo '' | gzip > ${prefix}.merged.fastq.gz" : ""
def touch_fail_fastq = (!save_trimmed_fail) ? "" : meta.single_end ? "echo '' | gzip > ${prefix}.fail.fastq.gz" : "echo '' | gzip > ${prefix}.paired.fail.fastq.gz ; echo '' | gzip > ${prefix}_1.fail.fastq.gz ; echo '' | gzip > ${prefix}_2.fail.fastq.gz"
"""
$touch_reads
$touch_fail_fastq
$touch_merged
touch "${prefix}.fastp.json"
touch "${prefix}.fastp.html"
touch "${prefix}.fastp.log"
cat <<-END_VERSIONS > versions.yml
"${task.process}":
fastp: \$(fastp --version 2>&1 | sed -e "s/fastp //g")
END_VERSIONS
"""
}
This generates the command to run fastp with 2 reads, where memory is allocated automatically.
#!/bin/bash -ue
[ ! -f GM12878_REP2_1.fastq.gz ] && ln -sf SRX1603630_T1_1.fastq.gz GM12878_REP2_1.fastq.gz
[ ! -f GM12878_REP2_2.fastq.gz ] && ln -sf SRX1603630_T1_2.fastq.gz GM12878_REP2_2.fastq.gz
fastp \
--in1 GM12878_REP2_1.fastq.gz \
--in2 GM12878_REP2_2.fastq.gz \
--out1 GM12878_REP2_1.fastp.fastq.gz \
--out2 GM12878_REP2_2.fastp.fastq.gz \
--json GM12878_REP2.fastp.json \
--html GM12878_REP2.fastp.html \
\
\
\
--thread 2 \
--detect_adapter_for_pe \
\
2>| >(tee GM12878_REP2.fastp.log >&2)
cat <<-END_VERSIONS > versions.yml
"FASTP_1":
fastp: $(fastp --version 2>&1 | sed -e "s/fastp //g")
END_VERSIONS
Configuration
This configuration allows the workflow to run with:
- Docker/Singularity for containerization
- Tracing and reporting capabilities
- Named processes for memory and CPU allocation
params{
outdir = "result"
trace_report_suffix = new java.util.Date().format( 'yyyy-MM-dd_HH-mm-ss')
input = "inputs/full_samplsheet.csv"
}
profiles {
singularity {
singularity.enabled = true
singularity.autoMounts = true
conda.enabled = false
docker.enabled = false
podman.enabled = false
shifter.enabled = false
charliecloud.enabled = false
apptainer.enabled = false
}
docker {
docker.enabled = true
conda.enabled = false
singularity.enabled = false
podman.enabled = false
shifter.enabled = false
charliecloud.enabled = false
apptainer.enabled = false
docker.runOptions = '-u $(id -u):$(id -g)'
}
emulate_amd64 {
// Run AMD64 containers on ARM hardware using emulation (slower but more compatible)
docker.runOptions = '-u $(id -u):$(id -g) --platform=linux/amd64'
}
}
singularity.registry = 'quay.io'
docker.registry = 'quay.io'
process{
withName:FASTQC_1{
cpus = 2
memory = 4.GB
}
withName:FASTQC_2{
cpus = 4
memory = 8.GB
}
withName:FASTQC_3{
cpus = 8
memory = 16 .GB
}
withName:FASTP_1{
cpus = 2
memory = 4.GB
}
withName:FASTP_2{
cpus = 4
memory = 8.GB
}
withName:FASTP_3{
cpus = 8
memory = 16 .GB
}
}
timeline {
enabled = true
file = "${params.outdir}/pipeline_info/execution_timeline_${params.trace_report_suffix}.html"
}
report {
enabled = true
file = "${params.outdir}/pipeline_info/execution_report_${params.trace_report_suffix}.html"
}
trace {
enabled = true
file = "${params.outdir}/pipeline_info/execution_trace_${params.trace_report_suffix}.txt"
}
Input Data
The sample sheet below includes two RNA-seq samples, each with input files ranging from 6 to 7 GB, representing real-world data sizes.
sample,fastq_1,fastq_2,strandedness
GM12878_REP1,s3://ngi-igenomes/test-data/rnaseq/SRX1603629_T1_1.fastq.gz,s3://ngi-igenomes/test-data/rnaseq/SRX1603629_T1_2.fastq.gz,reverse
GM12878_REP2,s3://ngi-igenomes/test-data/rnaseq/SRX1603630_T1_1.fastq.gz,s3://ngi-igenomes/test-data/rnaseq/SRX1603630_T1_2.fastq.gz,reverse
Running the Benchmark
The above scripts are stored in the GitHub repository: https://github.com/nttg8100/nextflow-cost-optimization
Clone the repository:
git clone https://github.com/nttg8100/nextflow-cost-optimization.git
cd nextflow-cost-optimization
- Requirements: At least 8 CPUs and 16 GB RAM (adjust based on your computing resources in
nextflow.config) - Alternative: If you don't have Docker, run with Singularity
Run the workflow, which will execute commands using Nextflow (installed via pixi). Docker is required on your machine.
# docker
make test-fastqc-docker-amd64
make test-fastp-docker-amd64
# if you use MacBook with ARM
make test-fastqc-docker-arm64
make test-fastp-docker-arm64
# if you do not have Docker installed, use Singularity
make test-fastqc-singularity
make test-fastp-singularity
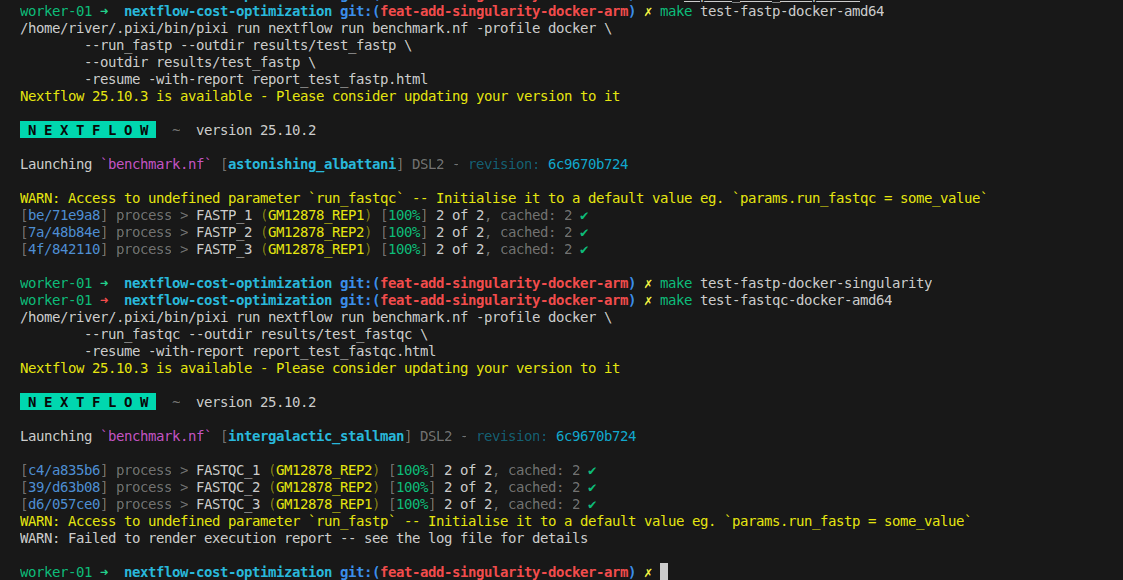
How to Evaluate Performance
What we need to optimize is finding the minimal CPUs and memory that can run tasks efficiently without being too slow. Open the HTML reports generated when you run the workflow to analyze performance. Below are the metrics we should use for evaluation:
-
%cpu:
The average percentage of CPU usage by the process during its execution. For multi-threaded jobs, this value can exceed 100% (e.g., 400% for a process using all 4 allocated CPUs fully). -
rss:
"Resident Set Size" — the maximum amount of physical memory (RAM) used by the process, measured in bytes. This shows how much memory your process actually consumed at peak. -
realtime:
The total elapsed (wall-clock) time taken by the process, from start to finish, including waiting and processing time.
These metrics help you tune and optimize your pipeline by revealing the true resource usage for each step.
FastQC
Interestingly, the execution time barely changes even when memory and CPUs are increased. From this experiment, we should consider using the minimal setup with 2 CPUs and 4 GB RAM while achieving nearly the same performance.
- Note: The module is taken directly from nf-core. There may be a bug related to FastQC that hasn't been identified yet.
- Finding: With real datasets, we can use minimal resources (much cheaper) without significant impact on running time!
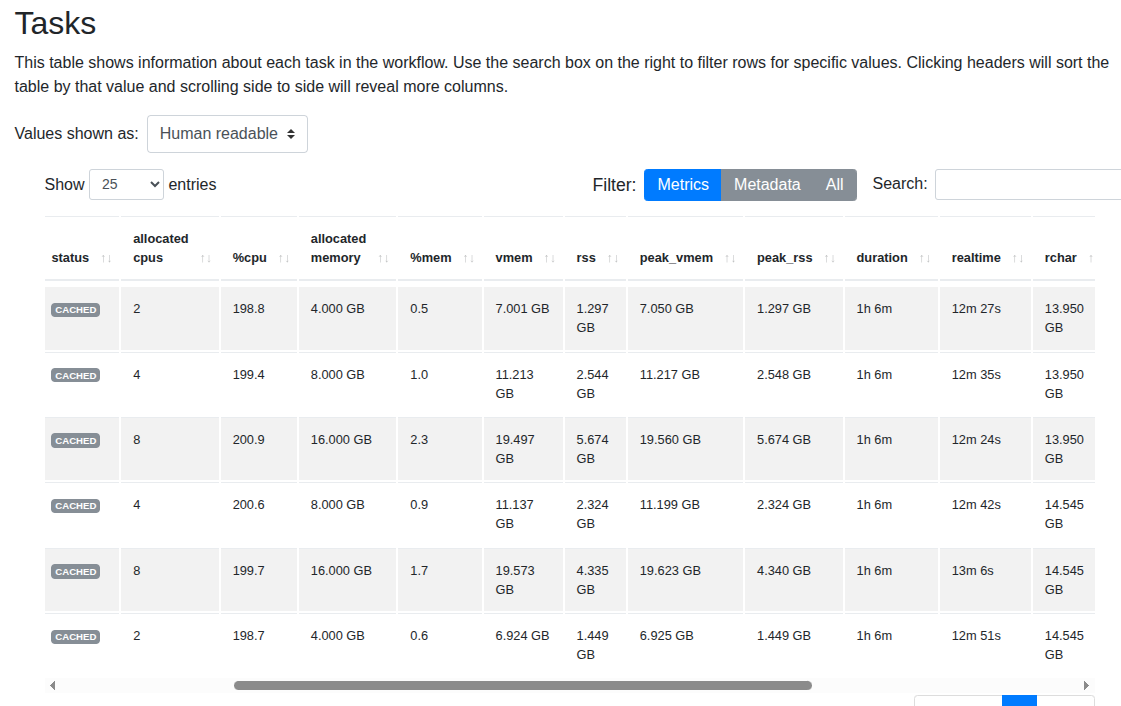
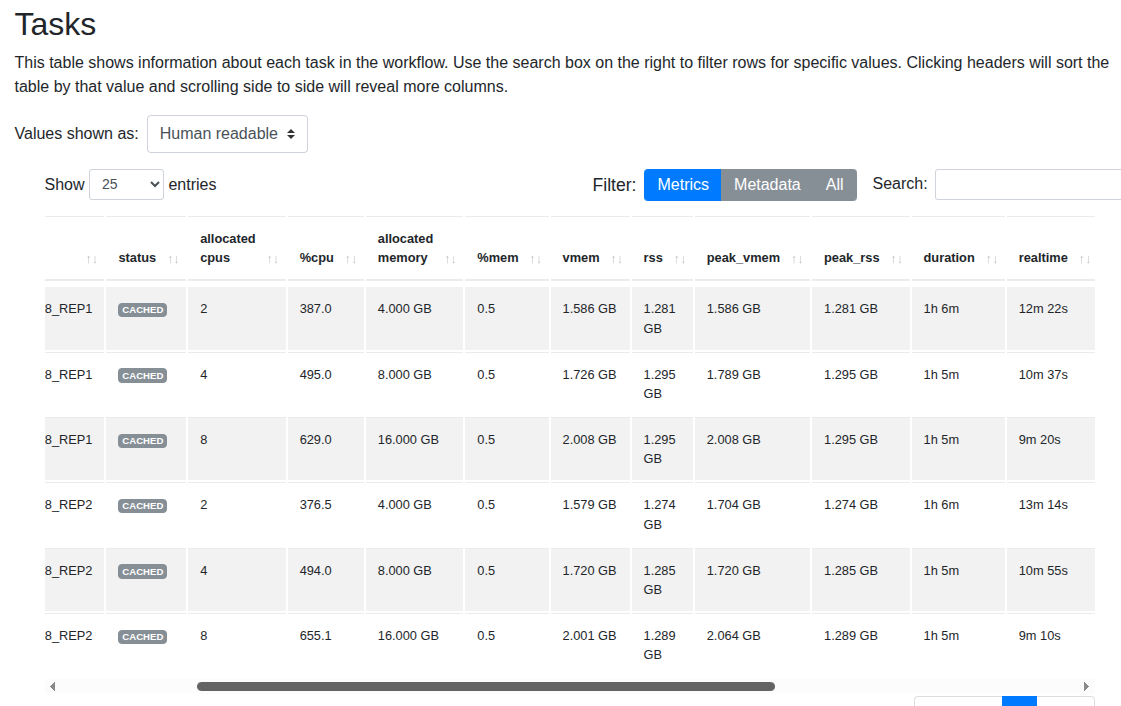
FASTP
With fastp, increasing memory and CPUs shows a slight reduction in running time.
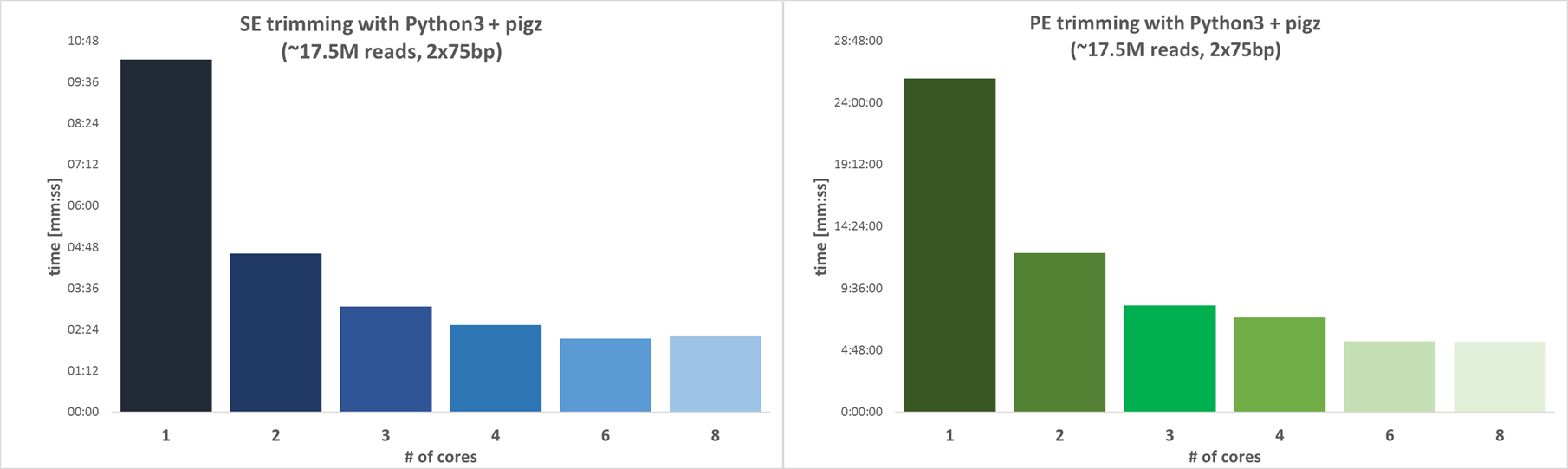
External benchmark
The simple example above can be modified appropriately for more tools. Here are external benchmarks showing that adjusting computing resources helps reduce running time while keeping costs nearly the same:
Trimgalore
Reference: https://github.com/FelixKrueger/TrimGalore/blob/master/CHANGELOG.md#version-060-release-on-1-mar-2019,
Using 4 CPUs is a good choice as it can reduce time by nearly 4x when compared with 1 CPU only.
STAR
Reference: https://academic.oup.com/bioinformatics/article/29/1/15/272537 Using 12 threads can align nearly 2x the reads when compared with 6 threads, while memory usage remains unchanged.
Nf-core application
- If you run workflow with HPC, consider the ratio between CPUs and memory. If you spend all of memory, there is no cpus left for another jobs
nf-core is one of the largest and most active bioinformatics workflow communities. However, their pipelines often use generic resource labels to define CPU and memory requirements for each process. For instance, the nf-core/rnaseq pipeline assigns default resources using labels like medium for tools such as FASTQC and FASTP.
Without tuning these settings for each specific tool, your workflow can waste resources and increase costs. In practice, both FASTQC and FASTP typically only need 2 CPUs and 4 GB RAM—much less than the default medium allocation.
This is especially important when running batch jobs in the cloud (AWS, Google Cloud, Azure, etc.), where virtual machines are often configured with a CPU:Memory ratio of 1:2. Requesting resources with a higher ratio, like 1:6, can increase queue times and lead to inefficient use of resources, as you may wait longer for an available instance and not fully utilize the extra memory.
withLabel:process_single {
cpus = { 1 }
memory = { 6.GB * task.attempt }
time = { 4.h * task.attempt }
}
withLabel:process_low {
cpus = { 2 * task.attempt }
memory = { 12.GB * task.attempt }
time = { 4.h * task.attempt }
}
withLabel:process_medium {
cpus = { 6 * task.attempt }
memory = { 36.GB * task.attempt }
time = { 8.h * task.attempt }
}
withLabel:process_high {
cpus = { 12 * task.attempt }
memory = { 72.GB * task.attempt }
time = { 16.h * task.attempt }
}
withLabel:process_long {
time = { 20.h * task.attempt }
}
withLabel:process_high_memory {
memory = { 200.GB * task.attempt }
}
To overwrite the existing worflow process cpus and memory, use the withName for specific tasks or adjust the label resources
process{
cpus = { 1 * task.attempt }
memory = { 2.GB * task.attempt }
time = { 4.h * task.attempt }
errorStrategy = { task.exitStatus in ((130..145) + 104 + 175) ? 'retry' : 'finish' }
maxRetries = 1
maxErrors = '-1'
withLabel:process_single {
cpus = { 1 }
memory = { 2.GB * task.attempt }
time = { 4.h * task.attempt }
}
withLabel:process_low {
cpus = { 2 * task.attempt }
memory = { 4.GB * task.attempt }
time = { 4.h * task.attempt }
}
withLabel:process_medium {
cpus = { 8 * task.attempt }
memory = { 16.GB * task.attempt }
time = { 8.h * task.attempt }
}
withLabel:process_high {
cpus = { 16 * task.attempt }
memory = { 32.GB * task.attempt }
time = { 16.h * task.attempt }
}
withLabel:process_long {
time = { 20.h * task.attempt }
}
withLabel:process_high_memory {
memory = { 32.GB * task.attempt }
}
withLabel:error_ignore {
errorStrategy = 'ignore'
}
withLabel:error_retry {
errorStrategy = 'retry'
maxRetries = 2
}
withLabel: process_gpu {
ext.use_gpu = { workflow.profile.contains('gpu') }
accelerator = { workflow.profile.contains('gpu') ? 1 : null }
}
withName: FASTQC{
cpus = { 2 * task.attempt }
memory = { 4.GB * task.attempt }
time = { 4.h * task.attempt }
}
withName: '.*:ALIGN_STAR:STAR_ALIGN|.*:ALIGN_STAR:STAR_ALIGN_IGENOMES'{
container = "docker.io/nttg8100/star:2.7.11b"
}
}
Lessons Learned
The two simple modules above help us select CPU and memory more easily. To optimize based on cost, here are some tips:
- For tasks shorter than 20 minutes, increasing CPUs and memory doesn't significantly reduce running time—use minimal resources instead
- Some tools benefit greatly from multiple CPUs and memory. For these, you should increase computing resources.
- Using 80% of available resources is typically sufficient. Sometimes when the input is slightly larger, tools can still run without crashing.