Bioinformatics Cost Optimization For Input Using Nextflow (Part 2)

Amazon S3 (Simple Storage Service) is built around the concept of storing files as objects, where each file is identified by a unique key rather than a traditional file system path. While this architecture offers scalability and flexibility for storage, it can present challenges when used as a standard file system, especially in bioinformatics workflows. When running Nextflow with S3 as the input/output backend, there are trade-offs to consider—particularly when dealing with large numbers of small files. In such cases, Nextflow may spend significant time handling downloads and uploads via the AWS CLI v2, which can impact overall workflow performance.On this blog post, we will start with downloading input first. Let’s explore this in more detail.
AWS CLI
Install
AWS CLI is the command line tools that helps work with AWS services. With nextflow, it can help for downloading inputs, uploading outputs. To install this tool and the tutorial on this blog, clone this repo. The repo uses the pixi and dokcer to quickly setup
git clone git@github.com:nttg8100/nextflow-cost-optimization.git
cd nextflow-cost-optimization
pixi shell
which aws
Explain Makefile, the below, we will run to start the docker service, upload files and the tar file of 10k files that can be used for benchmarking later
aws-config: start-minio
export AWS_ACCESS_KEY_ID="minioadmin"; \
export AWS_SECRET_ACCESS_KEY="minioadmin"; \
export AWS_DEFAULT_REGION="us-east-1"; \
export AWS_ENDPOINT_URL="http://localhost:9000" ; \
sleep 10 && aws s3 mb s3://io-benchmark --endpoint-url http://localhost:9000
results/tarball.tar:
@mkdir -p results/tarball
@count=10000; size=1M; index=1; \
for k in $$(seq $$count); do \
dd if=/dev/zero of=results/tarball/$${size}-$${index}-$$k.data bs=1 count=0 seek=$$size; \
done
tar -cvf results/tarball.tar -C results/tarball .
upload-tar:
aws s3 cp results/tarball.tar s3://io-benchmark/ --endpoint-url http://localhost:9000
upload-10k-files:
aws s3 cp results/tarball s3://io-benchmark/tarball --endpoint-url http://localhost:9000 --recursive
Start S3 service
Now you are ready to work with S3 object storage, this one will launch the minio, the simulated compatible S3 service with AWS. That will help to minimize the error related to your local computer and the remote bucket. This will create bucket called io-benchmark. Also simulated a lot of small files that we use later for proof of concept of this issue
make aws-config
To test the s3 service
# export env
export AWS_ACCESS_KEY_ID="minioadmin"
export AWS_SECRET_ACCESS_KEY="minioadmin"
export AWS_DEFAULT_REGION="us-east-1"
export AWS_ENDPOINT_URL="http://localhost:9000"
# configure threads
aws configure set default.s3.max_concurrent_requests 8
# test bucket
aws s3 ls --endpoint-url http://localhost:9000
# 1026-01-27 10:29:58 io-benchmark
Testing Download Independently
The above file content is the Makefile which simulates to create 1GB in total for a folder with 10k files, each file is 1MB. Nextflow is usually has the slow performance for input by 2 main reason:
- Verify download for each file
- Calculate inputs cache for all small files
It will create the additional tar file of these 10k files, that I will show you later why we create it and how it make your workflow easier
Without nextflow intervection, we want to test how long does it takes using aws cli v2 only
Download 10k files
Run this command to download
make upload-10k-files
for i in {1..3}; do /usr/bin/time -f "%e" aws s3 cp s3://io-benchmark/tarball ./tarball --endpoint-url http://localhost:9000 --recursive 2>&1 | tail -n 1; rm -rf ./tarball; done
The stderr shows that it takes around 110 seconds to download files
110.62
109.62
110.59
Download tarfile
Run this command to download, it will be much faster
make upload-tar
for i in {1..3}; do /usr/bin/time -f "%e" aws s3 cp s3://io-benchmark/tarball.tar . --endpoint-url http://localhost:9000 2>&1 | tail -n 1; rm -rf ./tarball.tar; done
The stderr shows that it takes less than 30 seconds to download this large file.
27.50
26.96
25.35
However, we need to have the small file inside, we can use pipe "|" to do it when we download file quickly
for i in {1..3}; do /usr/bin/time -f "%e" bash -c 'mkdir -p tarball && aws s3 cp s3://io-benchmark/tarball.tar - --endpoint-url http://localhost:9000 | tar -xvf - -C tarball' 2>&1 | tail -n 1; rm -rf ./tarball; done
The stderr shows that it takes less than 35 seconds to download and untar to get all small files
33.43
31.80
33.20
Fuse based system
Beside using the aws command to download individual files or tar file, using fuse based system is the alternative approach.
- For small files: FUSE enables applications to access individual files on demand without needing to download an entire archive or use complex commands, reducing overhead and making access fast and convenient.
- For large files: FUSE filesystems can fetch only the needed data chunks, allowing for efficient partial reads, sequential streaming, and avoiding unnecessary full downloads.
- General advantage: Since FUSE exposes cloud storage as a standard directory tree, workflows and tools that use local files work seamlessly, enabling parallel access and integration with caching and prefetching optimizations.
It can be used for using in the application that works with the large file or the folder contains many small files but it does not load entirely. For example, we annotate the variant for whole exome data but the annotation database is used for entire genome. We can use the tool to get only a few annotated variants region. Beside, distributed engine with fuse based system can be useful for distributed loading.
Makefile to quickly install mount-s3 via pixi. It will mount the whole bucket to the /mnt/vep_cache. This mount point is named after vep later it will be used direclty with VEP annotation example
mount-s3-vep-cache: ${HOME}/.pixi/bin/pixi
mkdir -p ./mnt/vep_cache
mkdir -p ./mnt/tmp
${HOME}/.pixi/bin/pixi run -e mount mount-s3 --endpoint-url http://localhost:9000 --region us-east-1 --force-path-style io-benchmark ./mnt/vep_cache --read-only --cache ./mnt/tmp --max-threads 8
Run this command to allow mount the remote s3 bucket
make mount-s3-vep-cache
# check
df
# Filesystem 1K-blocks Used Available Use% Mounted on
# udev 131907828 0 131907828 0% /dev
# tmpfs 26401936 3052 26398884 1% /run
# /dev/mapper/ubuntu--vg-ubuntu--lv 980760096 936881112 2913808 100% /
# tmpfs 132009668 0 132009668 0% /dev/shm
# tmpfs 5120 0 5120 0% /run/lock
# tmpfs 132009668 0 132009668 0% /run/qemu
# /dev/loop0 65408 65408 0 100% /snap/core20/2682
# /dev/loop1 65408 65408 0 100% /snap/core20/2686
# /dev/loop2 75776 75776 0 100% /snap/core22/2216
# /dev/loop3 75776 75776 0 100% /snap/core22/2292
# /dev/loop4 93696 93696 0 100% /snap/lxd/35819
# /dev/loop5 93696 93696 0 100% /snap/lxd/36918
# /dev/loop6 52224 52224 0 100% /snap/snapd/25577
# /dev/loop7 49280 49280 0 100% /snap/snapd/25935
# /dev/nvme0n1p2 1992552 433904 1437408 24% /boot
# /dev/nvme0n1p1 1098632 6228 1092404 1% /boot/efi
# controller-01:/home 476973568 349766144 106714624 77% /home
# tmpfs 26401932 4 26401928 1% /run/user/1000
# io-benchmark 1099511627776 0 1099511627776 0% /scratch/data/nextflow-cost-optimization/mnt/vep_cache
Now we can use this to simply download 10k files
for i in {1..3}; do /usr/bin/time -f "%e" bash -c 'cp -r ./mnt/vep_cache/tarball tarball ' 2>&1 | tail -n 1; rm -rf ./tarball; done
The stderr shows that it takes less than 45 seconds to download small files
44.94
46.06
45.46
Again, download only 10 GB tar file to see how it work
for i in {1..3}; do /usr/bin/time -f "%e" bash -c 'mkdir -p tarball && cp ./mnt/vep_cache/tarball.tar tarball' 2>&1 | tail -n 1; rm -rf ./tarball; done
The stderr shows that it takes less than 45 seconds to download small files
15.60
15.37
15.74
Again, download only 10 GB tar file to see how it work
for i in {1..3}; do /usr/bin/time -f "%e" bash -c 'mkdir -p tarball && cat ./mnt/vep_cache/tarball.tar|tar -xvf - -C tarball' 2>&1 | tail -n 1; rm -rf ./tarball; done
The stderr shows that it takes less than 45 seconds to download small files
19.68
21.86
20.31
Recap
Here is a summary table of the different approaches for downloading 10k small files (1GB total) from S3, with their typical performance and recommended use cases:
| Method | Download Time (s) | Pros | Cons | When to Use |
|---|---|---|---|---|
| AWS CLI (recursive, 10k files) | ~110 | Simple, no extra setup | Very slow for many small files | Rarely; only for small numbers of files |
| AWS CLI (tarball + untar via pipe) | ~33 | Fast, single download + extraction | Needs tar/untar logic | When workflow can handle tar extraction |
| FUSE (cp -r 10k files) | ~45 | Transparent, works like local FS | Needs FUSE setup, not always fastest | When random access or partial reads are needed |
| FUSE (cat tarball | tar -xvf -) | ~20 | Fast, combines FUSE and streaming untar | Needs FUSE setup, tar logic | For large archives with extraction |
Recommendations:
- For many small files, avoid direct recursive downloads; use tarballs or FUSE-based solutions.
- Use tarball + untar (via pipe) for best performance if you can bundle files.
- FUSE is ideal for workflows needing random access or partial reads, or when you can't change file structure.
- Nextflow (v25.04+) improves small file handling, but bundling or FUSE still offers significant gains for large numbers of files.
Nextflow integration
Workflow
We will simply run the workflow below that will accept the inputs from S3, download to use in this process and count the number of files inside
include { COUNT_FILES } from './modules/count_files_tar.nf'
include { COUNT_FILES_TAR } from './modules/count_files_tar.nf'
workflow {
main:
// benchmark files input
// normal files
if (params.benchmark_input){
ch_files = COUNT_FILES(Channel.fromPath(params.inputs).collect())
}
// tarball and untar
if (params.benchmark_input_tar){
ch_files = COUNT_FILES_TAR(Channel.fromPath(params.inputs).collect())
}
}
Modules
COUNT_FILES
It will accept a list of files directly from s3
process COUNT_FILES {
cpus 2
input:
path(file_path)
script:
"""
ls -lah tarball/**.data|wc -l > num_files.txt
"""
}
COUNT_FILES_TAR
It will download the tar file first, then inside the process untar later
For downloading file and use pipe to quickly untar will be applied later
process COUNT_FILES_ {
cpus 2
input:
path(file_path)
script:
"""
tar -xf ${file_path}
ls -lah tarball/**.data|wc -l > num_files.txt
"""
}
Remaining materials
It will includes the configuration with nextflow configs, include:
- nextflow.config: Standard config to run with docker, singularity and different platform
- nextflow_s3.config: S3 credential for minio storage
- nextflow_tar.config: The config that run with the local file later
These are the standard setup so I will not explain too much detail here, check at the below structure
├── benchmark_computing_resource.nf
├── benchmark_input.nf
├── inputs
│ ├── 1_samplesheet.csv
│ └── full_samplsheet.csv
├── LICENSE
├── Makefile
├── modules
│ ├── count_files.nf
│ ├── count_files_tar.nf
│ ├── fastp.nf
│ ├── fastqc.nf
│ └── vep.nf
├── nextflow.config
├── nextflow_s3.config
├── nextflow_tar.config
├── pixi.lock
├── pixi.toml
├── README.md
Testing
Using Makefile to simplify the process, it will run with different configuration. For proof of concepts, run once
test-input-standard: ${HOME}/.pixi/bin/pixi
${HOME}/.pixi/bin/pixi run -e standard nextflow run benchmark_input.nf \
-c nextflow_s3.config \
--benchmark_input \
--inputs="s3://io-benchmark/tarball"
test-input-tar: ${HOME}/.pixi/bin/pixi
${HOME}/.pixi/bin/pixi run -e standard nextflow run benchmark_input.nf \
-c nextflow_s3.config \
--benchmark_input_tar \
--inputs="s3://io-benchmark/tmp.txt"
Running with 10k files as input
Run the command below
time make test-input-standard
# 42,34s user 7,04s system 77% cpu 1:03,97 total
Running with 1 large file, untar in the process
time make test-input-tar
# 11,80s user 3,42s system 66% cpu 22,965 total
Magic improvement
Although we have show that we can use the single large tar file as input and download inside. However, we have to change the module. Is there any configuration can help ? Linking with previous section on using pipe after downloading, we can use this config
What it does it that it will use the same module while the files can be ingested using beforeScript
process{
withName:COUNT_FILES{
beforeScript = "mkdir -p tarball && aws s3 cp s3://io-benchmark/tarball.tar - --endpoint-url http://localhost:9000 | tar -xvf - -C tarball"
}
}
The command to run workflow, we can use a file to keep a place holder for nextflow to accept the input and also we add the config nextflow_tar.config
test-input-tar-pipe: ${HOME}/.pixi/bin/pixi
${HOME}/.pixi/bin/pixi run -e standard nextflow run benchmark_input.nf \
-c nextflow_s3.config -c nextflow_tar.config \
--benchmark_input \
--inputs="s3://io-benchmark/tmp.txt"
Remember to create the temp file and upload to the bucket
touch tmp.txt
aws s3 cp tmp.txt s3://io-benchmark --endpoint-url http://localhost:9000
Running the new setup
time make test-input-tar-pipe
# 11,33s user 2,85s system 82% cpu 17,166 total
Recap
- Using tar file and nextflow config can help reduce x3 times for downloading multiple small files as input
- We do not need to modify the existing module
Genomics England case study
Reference: https://aws.amazon.com/blogs/hpc/optimize-nextflow-workflows-on-aws-batch-with-mountpoint-for-amazon-s3/.
Issues
Now we can consider on what can be the issue that we can face with in the real problem. And how can we apply this Here, I found that the blog that how Genomic England can solve the similar issue when they want to use VEP to annotate their variants. The database that they used has many small files with 500GB in total. It will take more time to download data while takes a few minutes to annotate.
The pseudo code
process VEP {
input:
path(vcf)
val(vep_cache)
script:
"""
vep \
--input_file $vcf \
--fasta $params.human_reference_fasta \
--dir_cache $vep_cache
"""
}
Workflow and modules
Worflow is written simply to use only VEP module. Here to quickly reproduce, all parameters are hard coded
include { ENSEMBLVEP_VEP } from './modules/vep.nf'
workflow {
main:
ENSEMBLVEP_VEP(
Channel.of(tuple([id: 'HCC1395N'], file("inputs/vep_test_data.vcf.gz"), file("inputs/vep_test_data.vcf.gz.tbi"))), // tuple val(meta), path(vcf), path(custom_extra_files)
Channel.value("GRCh38"), // val genome
Channel.value("homo_sapiens"), // val species
Channel.value(114), // val cache_version
Channel.fromPath("s3://io-benchmark/vep_cache"), // path cache
Channel.of(tuple([:], [])), // tuple val(meta2), path(fasta)
Channel.of([]) // path extra_files
)
}
VEP module is collected from nf-core
process ENSEMBLVEP_VEP {
tag "${meta.id}"
label 'process_medium'
conda "${moduleDir}/environment.yml"
container "${workflow.containerEngine == 'singularity' && !task.ext.singularity_pull_docker_container
? 'https://community-cr-prod.seqera.io/docker/registry/v2/blobs/sha256/4b/4b5a8c173dc9beaa93effec76b99687fc926b1bd7be47df5d6ce19d7d6b4d6b7/data'
: 'community.wave.seqera.io/library/ensembl-vep:115.2--90ec797ecb088e9a'}"
input:
tuple val(meta), path(vcf), path(custom_extra_files)
val genome
val species
val cache_version
path cache
tuple val(meta2), path(fasta)
path extra_files
output:
tuple val(meta), path("*.vcf.gz"), emit: vcf, optional: true
tuple val(meta), path("*.vcf.gz.tbi"), emit: tbi, optional: true
tuple val(meta), path("*.tab.gz"), emit: tab, optional: true
tuple val(meta), path("*.json.gz"), emit: json, optional: true
path "*.html", emit: report, optional: true
path "versions.yml", emit: versions
when:
task.ext.when == null || task.ext.when
script:
def args = task.ext.args ?: ''
def args2 = task.ext.args2 ?: ''
def file_extension = args.contains("--vcf") ? 'vcf' : args.contains("--json") ? 'json' : args.contains("--tab") ? 'tab' : 'vcf'
def compress_cmd = args.contains("--compress_output") ? '' : '--compress_output bgzip'
def prefix = task.ext.prefix ?: "${meta.id}"
def dir_cache = cache ? "\${PWD}/${cache}" : "/.vep"
def reference = fasta ? "--fasta ${fasta}" : ""
def create_index = file_extension == "vcf" ? "tabix ${args2} ${prefix}.${file_extension}.gz" : ""
"""
vep \\
-i ${vcf} \\
-o ${prefix}.${file_extension}.gz \\
${args} \\
${compress_cmd} \\
${reference} \\
--assembly ${genome} \\
--species ${species} \\
--cache \\
--cache_version ${cache_version} \\
--dir_cache ${dir_cache} \\
--fork ${task.cpus}
${create_index}
cat <<-END_VERSIONS > versions.yml
"${task.process}":
ensemblvep: \$( echo \$(vep --help 2>&1) | sed 's/^.*Versions:.*ensembl-vep : //;s/ .*\$//')
tabix: \$(echo \$(tabix -h 2>&1) | sed 's/^.*Version: //; s/ .*\$//')
END_VERSIONS
"""
stub:
def prefix = task.ext.prefix ?: "${meta.id}"
def file_extension = args.contains("--vcf") ? 'vcf' : args.contains("--json") ? 'json' : args.contains("--tab") ? 'tab' : 'vcf'
def create_index = file_extension == "vcf" ? "touch ${prefix}.${file_extension}.gz.tbi" : ""
"""
echo "" | gzip > ${prefix}.${file_extension}.gz
${create_index}
touch ${prefix}_summary.html
cat <<-END_VERSIONS > versions.yml
"${task.process}":
ensemblvep: \$( echo \$(vep --help 2>&1) | sed 's/^.*Versions:.*ensembl-vep : //;s/ .*\$//')
tabix: \$(echo \$(tabix -h 2>&1) | sed 's/^.*Version: //; s/ .*\$//')
END_VERSIONS
"""
}
Their solution is showed below
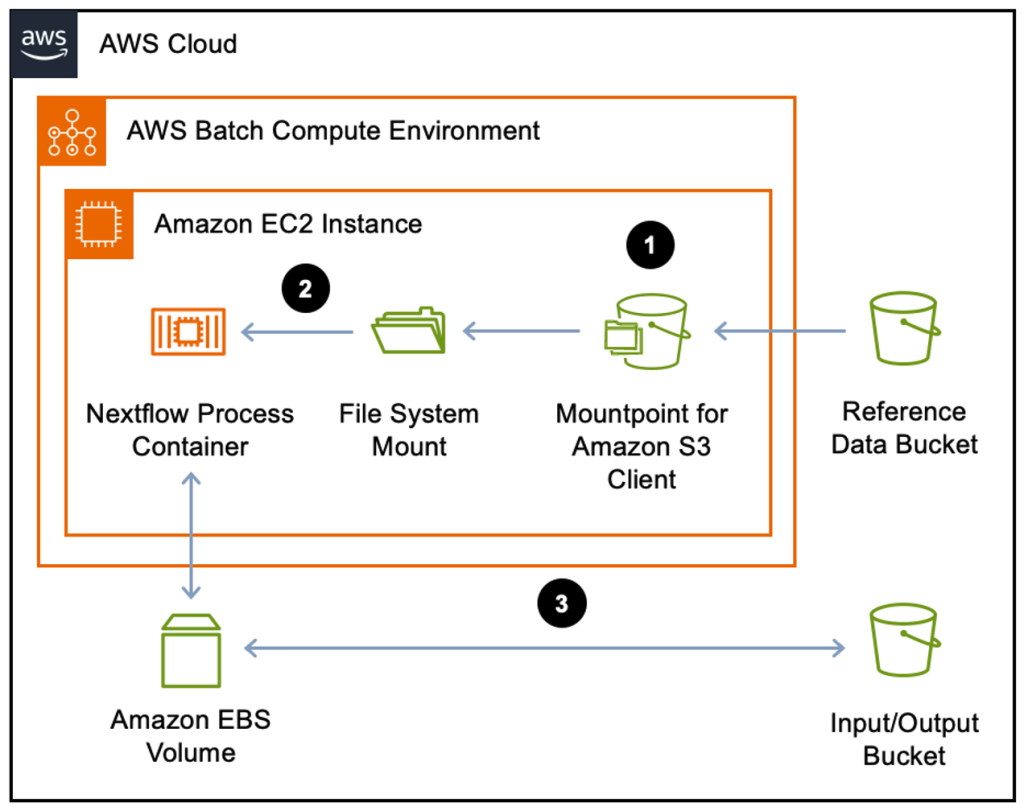
Inputs
The vcf file is prepared on the inputs folder, while the vep cache database (25.GB) can be prepared as below on Makefile.
vep/114_GRCh38: ${HOME}/.pixi/bin/pixi
mkdir -p vep
touch vep/vep_cache
cd vep && ${HOME}/.pixi/bin/pixi run -e core aws s3 --no-sign-request cp s3://annotation-cache/vep_cache/114_GRCh38 114_GRCh38 --recursive
vep/vep_cache.tar: vep/114_GRCh38
tar -cvf vep/vep_cache.tar -C vep/114_GRCh38 .
upload-vep-cache: vep/vep_cache.tar
${HOME}/.pixi/bin/pixi run -e core aws s3 cp vep/114_GRCh38 s3://io-benchmark/ --endpoint-url http://localhost:9000 --recursive
${HOME}/.pixi/bin/pixi run -e core aws s3 cp vep/vep_cache.tar s3://io-benchmark/ --endpoint-url http://localhost:9000
mount-s3-vep-cache: ${HOME}/.pixi/bin/pixi
mkdir -p ./mnt/vep_cache
mkdir -p ./mnt/tmp
${HOME}/.pixi/bin/pixi run -e mount mount-s3 --endpoint-url http://localhost:9000 --region us-east-1 --force-path-style io-benchmark ./mnt/vep_cache --read-only --cache ./mnt/tmp --max-threads 8
Simply run
# download and upload to local s3
make upload-vep-cach
# mount to local folder from local s3 via mountpoint-s3
make mount-s3-vep-cache
Benchmark
Here, I prepred the Makefile command to benchmark it quickly
test-vep-local: ${HOME}/.pixi/bin/pixi
${HOME}/.pixi/bin/pixi run -e core nextflow run benchmark_vep.nf -profile docker --vep_cache "./vep/114_GRCh38"
test-vep-mount-cache: ${HOME}/.pixi/bin/pixi
${HOME}/.pixi/bin/pixi run -e core nextflow run benchmark_vep.nf \
-c nextflow_s3.config -c nextflow_vep_mount.config --vep_cache "./mnt/vep_cache/114_GRCh38" -profile docker
test-vep-direct-s3: ${HOME}/.pixi/bin/pixi
${HOME}/.pixi/bin/pixi run -e core nextflow run benchmark_vep.nf \
-c nextflow_s3.config --vep_cache "s3://io-benchmark/114_GRCh38" -profile docker
test-vep-tar-cache: ${HOME}/.pixi/bin/pixi
${HOME}/.pixi/bin/pixi run -e core nextflow run benchmark_vep.nf \
-c nextflow_s3.config -c nextflow_vep_tar.config --vep_cache "s3://io-benchmark/vep_cache" -profile docker
test-vep-mount-tar: ${HOME}/.pixi/bin/pixi
${HOME}/.pixi/bin/pixi run -e core nextflow run benchmark_vep.nf \
-c nextflow_s3.config -c nextflow_vep_mount_tar.config --vep_cache "s3://io-benchmark/vep_cache" -profile docker
Now we can simply run the relative example test
make \<test-vep-benchmarl>
The following table summarizes the benchmark results for different approaches to accessing the VEP cache database during analysis:
| Method | Command | Time (min:sec) |
|---|---|---|
| Local cache | make test-vep-local | 2:26.20 |
| S3 mount (FUSE) | make test-vep-mount-cache | 2:31.08 |
| S3 mount tar (FUSE) | make test-vep-mount-tar | 5:57.18 |
| Direct S3 access | make test-vep-direct-s3 | 6:32.63 |
| S3 tar cache | make test-vep-tar-cache | 6:02.52 |
- As mentioned above, it does not need to download all of the database cache to annotate for the small vcf region files. That's why mounting the whole database is the best solution in this case
- Replacing the VEP command with
cp \$(readlink $cache) tmp -rin the module shows that it takes4:16.09to read from mount point and copy the whole folder to the new temporary folder. - Depended on your specific infrastructure, customize based on your need
Recap
- Using tar files and untarring large inputs does not always guarantee significant performance improvements. The benefit depends on the number of files and the total size of your input folder.
- If your tool only accesses a subset of the input files (rather than the entire folder), performance may be similar to having the database already available locally.
Recap
This comprehensive exploration of Nextflow's S3 integration revealed several key insights:
Performance Analysis
- Small files vs Large files: Nextflow handles large single files efficiently but struggles with many small files due to per-file operations
- Download methods: Tar files significantly outperform recursive downloads for small file collections (5x faster)
- FUSE benefits: Mount points provide transparent access with better performance and does not require large disk size for local storage.
Optimization Strategies
- Bundle small files: Use tar archives when dealing with numerous files (10k+ small files)
- Stream processing: Pipe extraction for immediate processing without full download
- Smart caching: Leverage FUSE-based caching for databases and reference data
- Choose the right tool: Select download method based on file count and access patterns
Practical Applications
- Genomics England case study: FUSE-based VEP annotation
- Bioinformatics pipelines: The principles apply to any workflow using remote data storage
- Cost considerations: Reduced download time + smaller disk size = lower compute costs on cloud platforms.
Key Takeaways
- Profile before optimizing: Measure actual bottlenecks in your specific workflows
- Consider file distribution: Many small ≠ few large when choosing download strategies
- Use Nextflow v25.04+: Improved small file handling reduces need for workarounds
- Leverage FUSE: Provides transparent access with better performance than CLI tools
- Design for scale: Build workflows that work efficiently with your data characteristics
Whether you're working with thousands of small files or terabytes of reference data, understanding these patterns helps you design more efficient, cost-effective bioinformatics workflows.